Post provided by Bruno do Rosario Petrucci
Inferring rates of diversification from phylogenies and fossils has been a focus of evolutionary biology for generations, since a quantitative understanding of the dynamics of speciation and extinction is necessary for a complete picture of the history of life. Computational and statistical methods with that goal have recently seen an explosion in complexity and power. While exciting, this exploration needs to be accompanied by exhaustive testing, and I have been motivated by such work since my undergraduate days.
The birth of an idea
This project started with an e-mail to Prof. Tiago Quental, at the University of São Paulo. After two years in physics, and a year in eco-evolutionary theory, I decided macroevolution was a great field to apply my quantitative skills while investigating some interesting questions about nature. Tiago not only took me on, allowing me to spend my last summer before graduation in my hometown, but also pointed me towards amazing researchers at my own school, the University of Chicago. This led me to my undergraduate thesis advisor, Prof. Michael Foote and Prof. Graham Slater, among many others. Ultimately, this led directly into my PhD, since all my mentors at UChicago recommended I apply to my current advisor, Prof. Tracy Heath. The first signs that this was the line of work for me, however, came from the work in Tiago’s lab itself.
Initially, the plan was to test the accuracy of PyRate, a popular software for inferring diversification rates from fossil records. To conduct these tests, I needed to simulate species diversification. That’s how one commonly would test methods of inference—simulate data under certain parameters, compare the simulation parameters to the ones inferred, and then gain some insight on the strengths and weaknesses of the method. Since macroevolution is a historical science, simulating data is the only way we can get data for which we know the true generating parameters. By varying the complexity of my simulations, we can thoroughly gauge the robustness of a given model.
Throughout this work, however, I became somewhat frustrated with the available tools for simulation in the field. Many powerful software lacked some simulation scenarios I wanted to test, and to run simulations across my desired range of parameters, I would need to use multiple packages, with multiple different implementations, output organization, etc.
Amidst this conflict, I had a number of stimulating conversations with another member of Tiago’s lab: Matheus Januário, now a PhD candidate under Prof. Daniel Rabosky. His experience with R was essential to develop our ideas on efficient diversification simulation. He also possesses a biological intuition that complemented my quantitative background, especially since I was still an uninitiated biologist. Though, his quantitative skills were nothing to scoff at—he was working as Tiago’s computational assistant—and his experience with R was essential to develop our ideas on efficient diversification simulation.
After many lunches and coffee breaks worth of math and programming discussions, and lots of varyingly successful attempts at implementing our ideas, we pitched our proposal to Tiago.
Tiago is nothing if not thorough, and justifiably made us do our due diligence. After we managed to convince him that this would be a valuable addition to the field, my summer project took a turn. We’re writing an R package! paleobuddy was born.
paleobuddy
To understand why we embarked on this journey, let me pitch paleobuddy to you as we did to Tiago three years ago. Most simulation packages in the field have some trouble handling rates of diversification that vary with time. If rates are constant, we just need to use the rexp() R function to draw times of speciation and extinction from an exponential distribution—this process is called a birth-death process. If they depend on time, however, packages vary in the way they handle this issue, but the solution is usually rather slow. By generalizing the rexp() R function to allow for time-dependent rates, we are able to simulate continuous time dependence faster than almost any other package. We further generalized rexp() to follow the Weibull, a distribution frequently used to model age-dependent diversification, i.e. speciation or extinction probability changing with age. We can even combine those two by varying the Weibull parameters with time, which was one of the scenarios that first led me to start writing what later became paleobuddy.
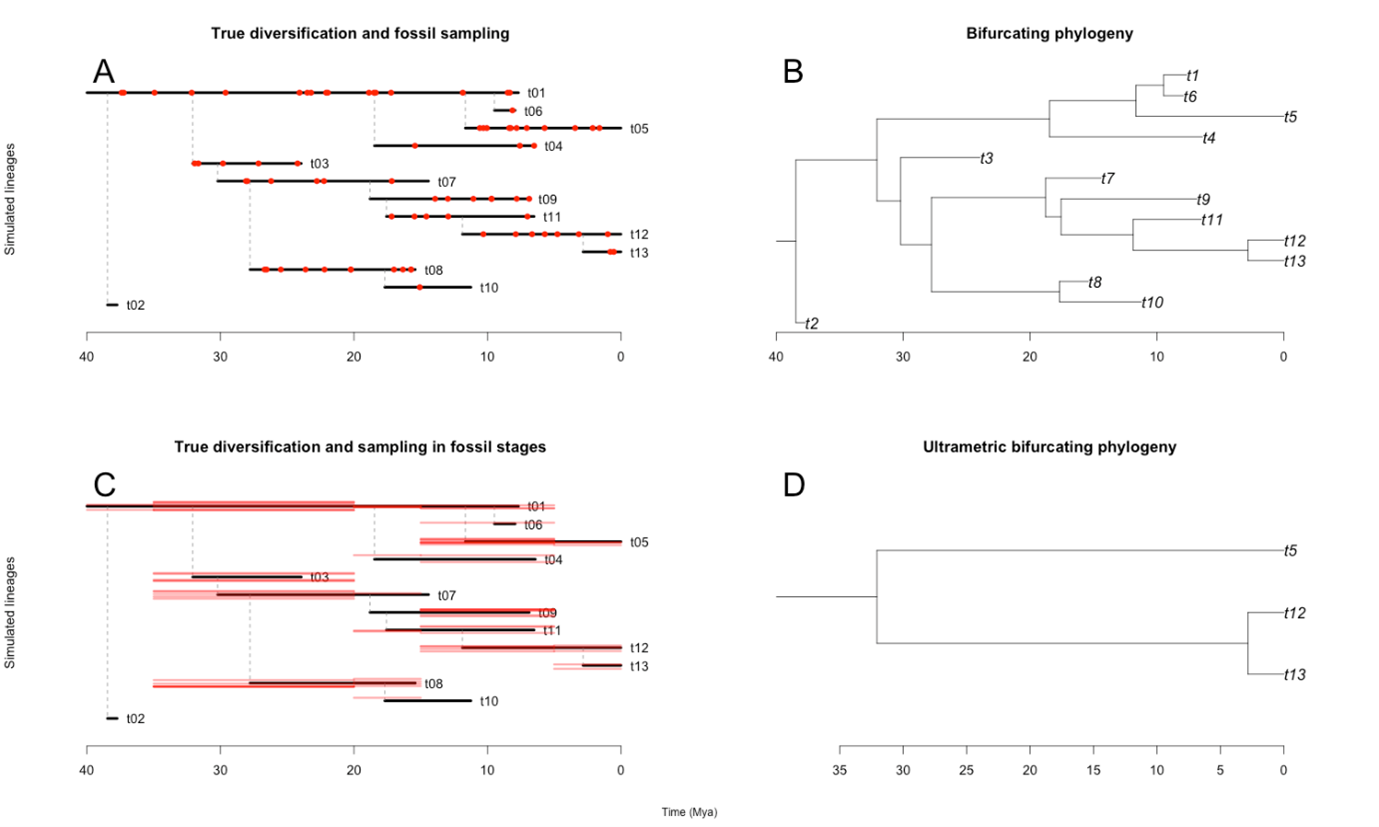
While this innovation enabled paleobuddy to have a number of simulation scenarios previously unavailable (see table below), the framework built around generalizing rexp() also has its own benefits. First, expanding the range of simulation scenarios is straightforward. For instance, I have recently implemented trait-dependent diversification (see dev_traits branch in the github) by simply translating the process into a modification of our rexp() custom function. Furthermore, since fossil sampling is modeled in a similar way, we can transfer all the flexibility in our diversification simulations to sampling strategies. paleobuddy is therefore flexible, easily expandable, and able to output both fossil records and phylogenetic trees from the same process (see figure below). Hopefully it’s understandable why we decided to abandon the initial idea for that project and focus on the package.

But do you use it?
paleobuddy became a substantial part of my research from then on. I made its development the focus of my undergraduate honors thesis at UChicago, which allowed me to incorporate many insights from my mentor, Prof. Michael Foote. The package was the topic of my first poster presentation, and my first research talks. I still do the vast majority of simulations for my PhD research in paleobuddy, and our focus on expandability paid off as the range of models I study keeps growing. Tiago’s guidance on exhaustively documenting the package—mostly inspired by another powerful simulator in the field, paleotree-–has also paid off whenever I go back to modify functions I haven’t looked at in years. The package is a central part of my research, and for good reason. We have seen theoretical macroevolution expand at a rapid pace, and that requires tools for validation to keep up. An expandable, efficient simulator, such as paleobuddy, is a valuable contribution to this endeavor.
In the three and a half years since we started working on it, paleobuddy shaped the scientist I am today. Developing it was a crash course in how we do theory in macroevolution, and the problem-solving skills it has forced me to hone can sometimes feel like superpowers. It’s wild to think I’ve been working on it and using it for as long as I’ve been an evolutionary biologist. It does feel like, in some philosophical sense, paleobuddy is—up to now, at least—my academic career. It has been with me since I arrived, and I plan to keep it going while I’m here. Maybe that’s why I decided on that name. More than a pillar of my academic career, and a part of my contribution to the field, maybe in some emotional sense I see this package as a comfortable friend amidst the hardships of academia. No matter the obstacles the job throws at me, I can always go back to my little bundle of R code, and all the friends it has brought into my life.
Click here to read the full article “paleobuddy: An R package for flexible simulations of diversification and fossil sampling” (https://doi.org/10.1111/2041-210X.13996)
