Post provided by Sandra J. Simon
Working with a Genetic Model
During my PhD at West Virginia University (WVU), I worked with the genetic models in the family Salicaceae, such as Populus trichocarpa,to understand the relationship between plant genetics and biotic interactions. Let’s take a moment to focus on what makes P. trichocarpa a good species to use as a genetic model by comparing it to the human genome! As you might remember from general biology, humans have 23 pairs of chromosomes and the human reference genome contains ~3.2 billion base pairs (bps). Scientists began sequencing the human genome in 1990 and it took researchers 13 years to finish sequencing that entire genetic code! Today, sequencing technology has greatly improved, allowing us to generate all of that information in under 24 hours – over 4700 times faster.

P. trichocarpa’s genome has 19 pairs of chromosomes and its reference genome is much smaller than ours at ~410 million bps. Due to its compact size, P. trichocarpa is very compatible with modern sequencing technology making it an easy species to use for genetic research. To say that this simplified my work as a graduate student would be a drastic understatement, but all of the researchers not working on genetic model organisms are not so lucky. Most organisms have far more complex genomes, making it extremely difficult to produce a full genome sequence with current technology. Study of these organisms is further complicated by difficulty sampling
The Woes of Genetic Sequencing
In a perfect world our extractions would always yield DNA that is ready to throw on a PacBio sequencer. In reality, most extractions are done with small amounts of non-ideal tissue that result in degraded DNA at low quantities. In the early brainstorming sessions developing ISSRseq we wanted to have it all: a technique that uses minimal amounts of DNA, can be used on many different species, has flexible pipelines for bioinformatics and data analysis, and is user friendly. If you have found yourself in a similar position and the list above sounds great then read on!
ISSRseq: How it Works
ISSRseq is a polymerase chain reaction (PCR) based method which requires only small amounts of DNA and can be used with degraded samples. It targets regions known as inter simple sequence repeats (ISSR) to sequence portions of the genome and generate single nucleotide polymorphism (SNP) data. We used the genetic model Populus deltoides to show that sequenced loci are located throughout the genome and not clustered on particular chromosomes. We then tested ISSR primers in a scenario more applicable to other researchers by using two non-model orchid species, Corallorhiza bentleyi and C. striata. This showed that ISSRseq is capable of generating SNP datasets that are comparable with existing yet more technologically demanding and complicated reduced representation sequencing methods (RRS).
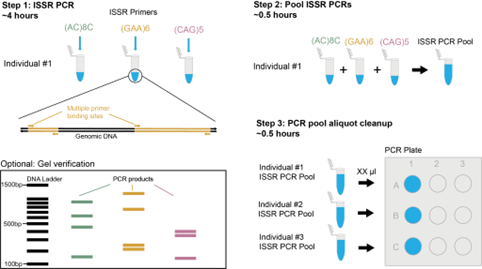
Although we focused on three plant species in our manuscript, the most promising impact of ISSRseq lies in its simplicity. Due to the prevalence of simple sequence repeats (SSRs) in genomes it could be used to not only study any biological organism but symbiotic or pathogenic relationships across multiple organisms simultaneously!
Making ISSRseq User Friendly
As a graduate student I worked with the supervisors of the WVU undergraduate biology capstone to write protocols to use for student genetic experiments. One of my most vivid memories was helping out with a wet-lab and hearing one undergraduate say “Who wrote this protocol?! It sucks!”. Four semesters of that gave me a lot of time to revise and test our protocols with new users. Coupling that experience with assisting our research undergraduates (and now coauthors Nicole and Mathilda!) as they generated data for the ISSRseq manuscript allowed us to fully adapt it into our capstone class as a research module for new students.

We have continued to work on helping new users by developing additional online resources. You can now find our website that has videos discussing fungal amplicon sequencing and ISSRseq and a GitHub page devoted to bioinformatics and analysis (see additional links below)!
The Possibilities of ISSRseq
What we found during our study merely scratches the surface of the utility of ISSRseq in genetic and genomic research. For example, flexibility in the bioinformatic pipeline allows researchers to identify large structural mutations such as insertions and deletions (indels). Simple modifications of the molecular method conditions could also amplify loci that are thousands of base pairs in length, greatly improving our understanding of non-model genomic structure. With the tools we have developed, ISSRseq provides flexibility for more experienced researchers and a clear introduction to both students and new labs to the field of population genetics and phylogeography.
Additional Links
To read the full paper, ‘ISSRseq: An extensible method for reduced representation sequencing’, click here. (https://doi.org/10.1111/2041-210X.13784)
To access videos discussing fungal amplicon sequencing, click here. (https://www.invasiongenomics.com/dna.html)
To visit GitHub wiki for wet-lab, bioinformatics and data analysis protocols, click here. (https://github.com/btsinn/ISSRseq/wiki)
