Post provided by Marco Crotti
How organisms adapt to the environment they live in is a key question in evolutionary biology. Genetic variation, i.e. how individuals within populations differ from each other in terms of their DNA, is an essential element in the process of adaptation. It can arise through different mechanisms, including DNA mutations, genetic drift, and recombination.
Differences in DNA sequences between individuals can results in differences in the expression of genes. This can therefore determine the organism’s capacity to grow, develop, and react to environmental stimuli. However, a growing body of literature reveals that there are other ways organisms can change the way they interact with the world without mutations in the DNA sequence.
Epigenetic Mechanisms
Epigenetics refers to the molecular processes that influence the expression of genes, and therefore the phenotype (i.e. set of observable characteristics), of organisms without actually changing the DNA sequence of those genes.
Within the last decade, there has been growing research interest in these mechanisms. Epigenetic mechanisms can explain the process of trait variation, rapid phenotypic differentiation, and adaptation to new environments in the absence of genetic differentiation. The most common, or at least most studied, epigenetic mechanism is DNA methylation, which involves the addition of a methyl group to cytosine, one of the building blocks of DNA.
Current Methods for Studying DNA Methylation
While it would be ideal to combine multiple types of DNA data (DNA sequences and DNA methylation), to better understand the relationship between the phenotype and genotype, this approach has not been used much to date. This is mainly due to the high cost of current sequencing technology. Ideally, you would need a method to obtain both nucleotide sequences and DNA methylation information simultaneously, rather than spending lots of money and time on two separate methods.
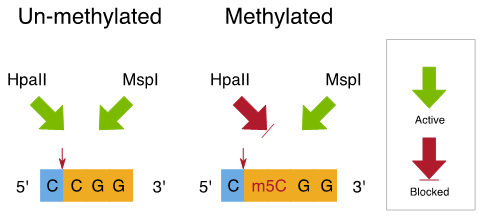
Among the many techniques available to study DNA methylation, epigenetic Restriction site Associated DNA sequencing (EpiRADseq) stands out for its simplicity, power, and low cost. This is based on a well-known technique called double digest Restriction site Associated DNA sequencing (ddRADseq), which focuses on a relatively small number of nucleotide sequences from all across the genome to study DNA variation.
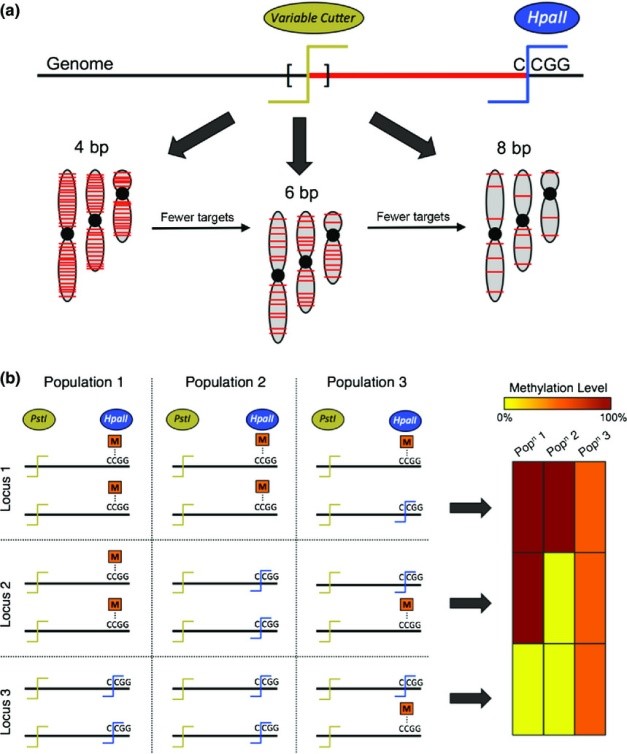
EpiRADseq and ddRADseq techniques work by using two restriction enzymes that cut apart the DNA into small fragments for sequencing a reduced representation of the genome. The only difference between the two methods is that in EpiRADseq, one of the restriction enzymes has to detect methylated cytosine. By comparing loci that are present in some individuals and not others, we can infer methylation and its variation across individuals, populations, or treatments. However, the underpinning genetic variation is also important to know because it can affect methylation signals. This means that best practice is to generate both epigenetic and genetic information to study populations.
Combining Genetic and Epigenetic Data Analysis
Because EpiRADseq and ddRADseq are so similar, we thought about testing whether data from EpiRADseq, originally developed for DNA methylation analysis only, could also be used to study DNA variation. This would ultimately cut the consumable and sequencing costs as well as the bench time required in half compared to generating data for both separately.

To test this approach, we used a previously published dataset on Porites corals that had used ddRADseq and EpiRADseq for each individual, and we generated ddRADseq and EpiRADseq data for European whitefish from Scotland. These two study organisms are deeply divergent, being an invertebrate and a vertebrate, but both are valuable exemplars for ecology and evolution studies.

After deciding on these two datasets, we then ran some standard population genetic analyses, and compared the estimates obtained from ddRADseq and EpiRADseq. We found that the number of SNPs (single nucleotide polymorphisms) was very similar between the two molecular methods. We determined that there was a strong correlation between methods for summary genetic statistics such as level of heterozygosity and nucleotide diversity. We also found that identical patterns of population genetic structure were recovered using SNPs from EpiRADseq and ddRADseq approaches.
Summary
Overall, our results show that it is possible to use one technique, EpiRADseq, to study both DNA variation and DNA methylation. This new perspective is helpful because it allows researchers to save time and money by using a single technique for two types of analysis and combine different level of DNA information to get a better idea of the processes behind response and adaptation.
To read more about using epigenetic RAD sequencing to study DNA variation and DNA methylation, check out the Methods in Ecology and Evolution article, ‘Population genomic SNPs from epigenetic RADs: gaining genetic and epigenetic data from a single established next‐generation sequencing approach’
