
Post provided by Nyniane Steinkampf–Pellecuer, Idriss Pelletan and Pauline Provini
We are two PhD students and a researcher at the MECADEV lab of the Muséum national d’Histoire naturelle of Paris. As part of our research on birds’ evolution, we study the functional morphology of their organs to understand how their shape is linked to their function, and how they evolved. Due to the size of the group we study (birds have more than 11,000 species) and other constraints such as specimen availability in the collections, we must conduct our analyses on subsets of species. From our results, we can then infer evolution at the level of the entire group. However, to ensure the robustness of these analyses and results, our species samples must be representative of the group we are studying, and more particularly of its evolutionary history, i.e., its phylogeny.
As we were discussing together, and after reviewing the scientific literature, it appeared that there was no method to ensure the phylogenetic representativeness of a taxonomical sample. The common practice consisting of increasing sample size to increase phylogenetic coverage didn’t seem reliable. There is a real methodological gap here, because the creation of a good sample is crucial to ensure the quality of a study. However, until now this step is not standardized and is often neglected.
On a blocked train, Nyniane had the idea of using Faith Phylogenetic Diversity index to quantitatively measure the phylogenetic quality of her sample by comparing it to random samples of the same size. This approach offered a direct and simple way to evaluate the phylogenetic representativeness of a sample. After discussing the idea together, we realized the potential of the method. We found that we could extend the method to the design of optimized samples, based on a suggestion made by Faith in his original publication, and never followed until now. Pauline also had the idea to create an R package to make this promising method available to all.
We didn’t have much experience in creating R packages, and after a long work of translating our ideas into R functions, we were able to create a package that we called SOUPE for “Sample Optimization Using Phylogeny and Ecology”. This package is available in open access to all the scientific community on our GitHub page (https://github.com/BirdsongTeam/Supplementary-Material-Steinkampf–Pellecuer-Pelletan-Provini-SOUPE). To facilitate user’s implementation of the method, we have created two processes (Figure 1). The first one is the a posteriori process. It evaluates the phylogenetic quality of a sample by comparing it to randomly generated samples of the same size. The second one is the a priori process. It builds samples that are optimized for phylogeny through an iterative algorithm that maximizes their Faith index. Both processes come with various options such that data availability restrictions, consideration of key species, ecological parameters optimization, in order to meet study requirements.
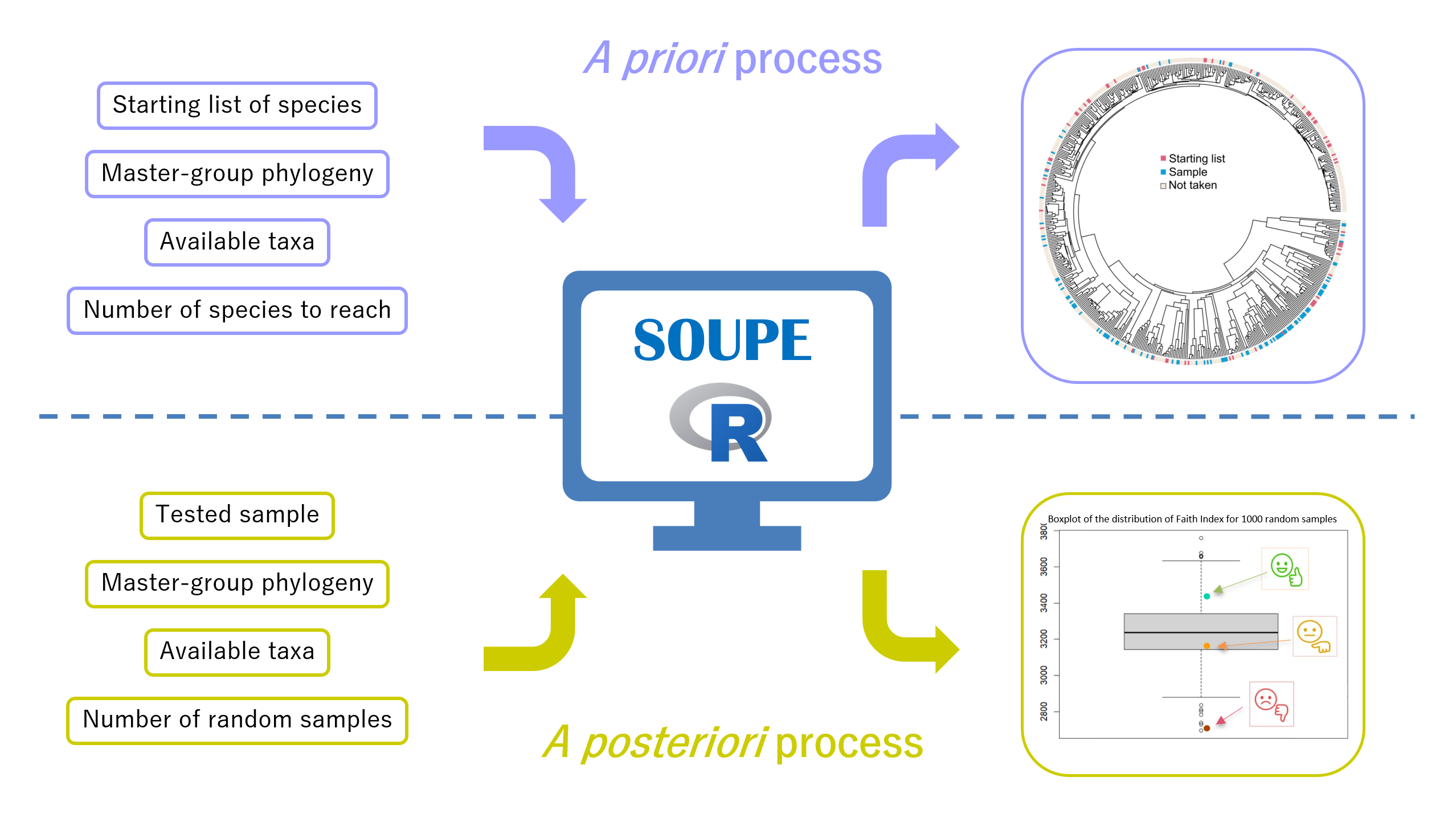
Easy to use, the package is accompanied by a detailed tutorial and a precise decision tree to navigate through the different functions and options of the method. It requires few files as input and is adaptable to all kinds of questioning and taxa. SOUPE method offers a reproducible, objective and quantitative way to test the phylogenetic quality of a sample, or to create representative samples, while taking into account the needs of each study. It will allow researchers to save time, reduce sample size, and most importantly, it will guarantee the phylogenetic quality and robustness of their results.
Read the article here.
