Post provided by Ruben Props, Michelle Berry, Marian Schmidt, Frederiek-Maarten Kerckhof, Vincent Denef and Nico Boon

Exploring microbial diversity and relating it to ecosystem functions is one of the primary occupations of microbiologists and microbial ecologists worldwide. Unfortunately, recent studies have shown that the microbial census is far from complete and that it is heavily biased towards certain (host-associated) environments. With the Earth’s microbial diversity estimated at an impressive one trillion (1012) taxa, the search continues for new technologies and methodologies that may help us better describe, monitor and preserve the microbial diversity of our planet’s natural and engineered ecosystems.
The Challenge of Estimating Microbial Diversity
Since Carl Woese’s landmark paper in 1977 demonstrated that marker genes, such as the 16S rRNA gene, could be used to classify bacterial and archaeal taxa, assessment of microbial diversity has increasingly relied on molecular technologies. This laid the conceptual foundation for many sequence-based microbial diversity measurements.
Nowadays, two major sequence-based approaches are used:
- The standard: Operational Taxonomic Units (OTUs)
Here, microbial diversity is estimated by grouping similar sequences of marker genes. A specified sequence similarity cut-off (usually of 97%) serves as an operational definition of a prokaryotic species, known as an operational taxonomic unit or OTU. Based on the distribution of these OTUs, diversity estimates can be calculated.
- Additional refinement: Oligotyping
Often single OTUs conceal multiple ecologically distinct taxa, which can sometimes be identified by more refined computational tools, such as oligotyping. Oligotyping has been shown to differentiate marker sequences that are more than 97% similar and can potentially reveal populations with ecologically distinct characteristics. For example, oligotyping was used to distinguish taxa in the human oral microbiome with different habitat preferences (e.g. throat, tongue, and saliva) that could not be resolved at the OTU level.
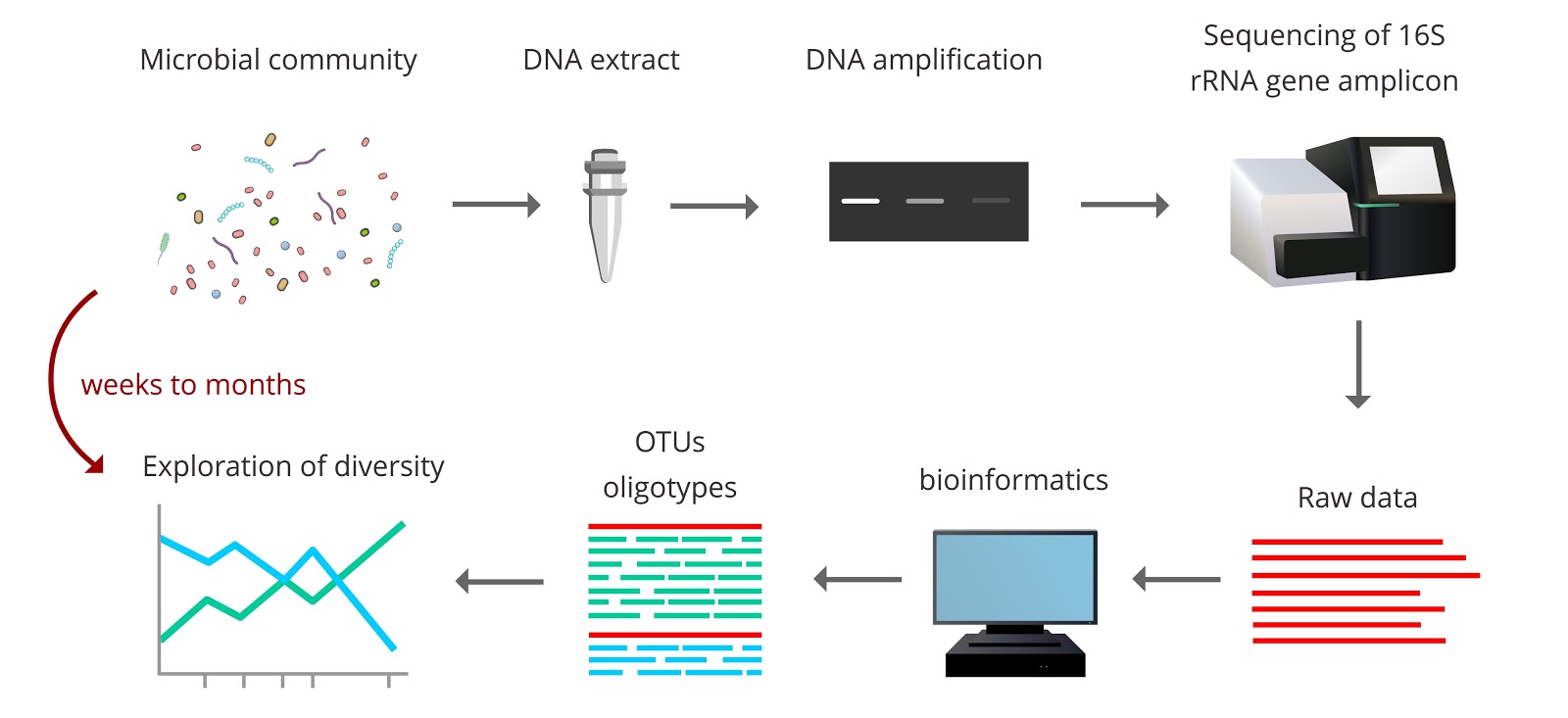
Despite the fact that these sequence-based approaches are considered high-throughput molecular technologies, they remain laborious, slow and costly for extensive sampling campaigns. It is not uncommon for it to take months to get from the initial sample collection to the final diversity estimates.
Due to these constraints, environmental sampling campaigns are nearly always performed under “black box” sampling designs. This means that environmental samples are taken with little to no prior knowledge of whether the sampling strategy will yield an accurate representation of the microbial diversity present in the environment. While the obvious solution to this issue would be to simply increase the amount and frequency of samples taken, budget and time restraints usually limit this approach.
The (Partial) Solution: Single-Cell Measurements
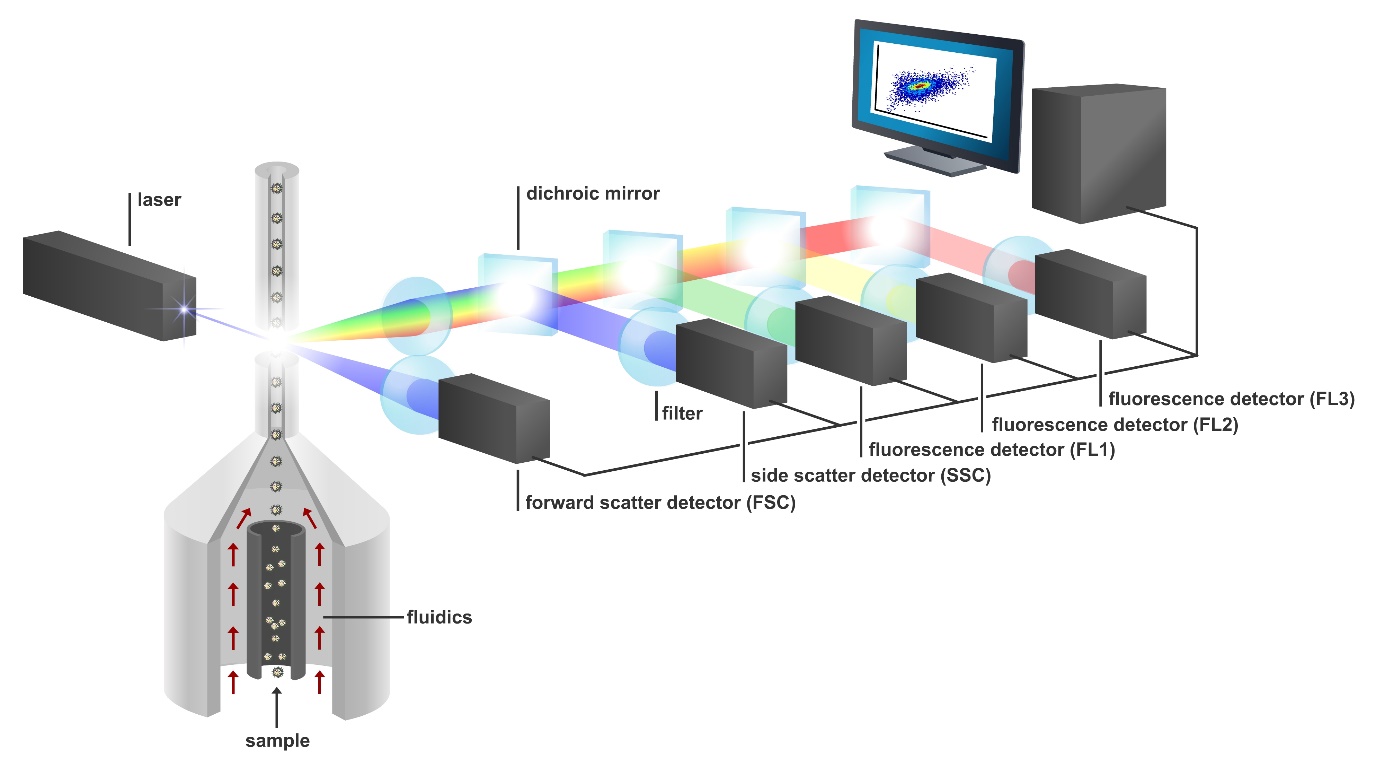
Our research group developed a new method to inexpensively and rapidly estimate microbial diversity based on high-speed identification of phenotypic features of microbes. Central to our approach is a technique called ‘flow cytometry’, which shoots individual microbial cells through a high-powered laser beam at rates of up to 50,000 microbes s-1, while simultaneously registering their scattered and emitted light on multiple detectors. Microbes with a different morphology (think about cell size, shape and density) induce different scatter effects. Because we also dye the microbes with a fluorescent stain, microbes that differ in DNA or RNA content will emit different fluorescence signals.
Ultimately, our hypothesis was that these phenotypic single-cell signals would resolve ecologically distinct taxa, allowing us to translate the multidimensional data patterns into robust diversity estimates, called phenotypic diversity metrics. Conceptually, many similarities exist between our approach and that of the recently validated acoustic indices for macroscopic marine ecosystems, where ecosystem diversity was estimated based upon the organisms’ sound patterns.
In a second stage of our work, we investigated the performance of these phenotypic diversity metrics for a freshwater ecosystem and validated them with the ‘gold standard’ sequence-based approaches. The ecosystem in question, a freshwater basin, was part of the secondary cooling water circuit of a nuclear test reactor. We anticipated a well-defined shift in diversity to occur, as the reactor start-up induces an abrupt perturbation in temperature and chemistry of the ecosystem. As it turns out, our phenotypic diversity metrics indicated a severe diversity shift taking place within the first few hours of operation. Based on the phenotypic diversity profiles, we selected samples for sequence-based analysis, which – to our excitement – confirmed that these diversity shifts were real.
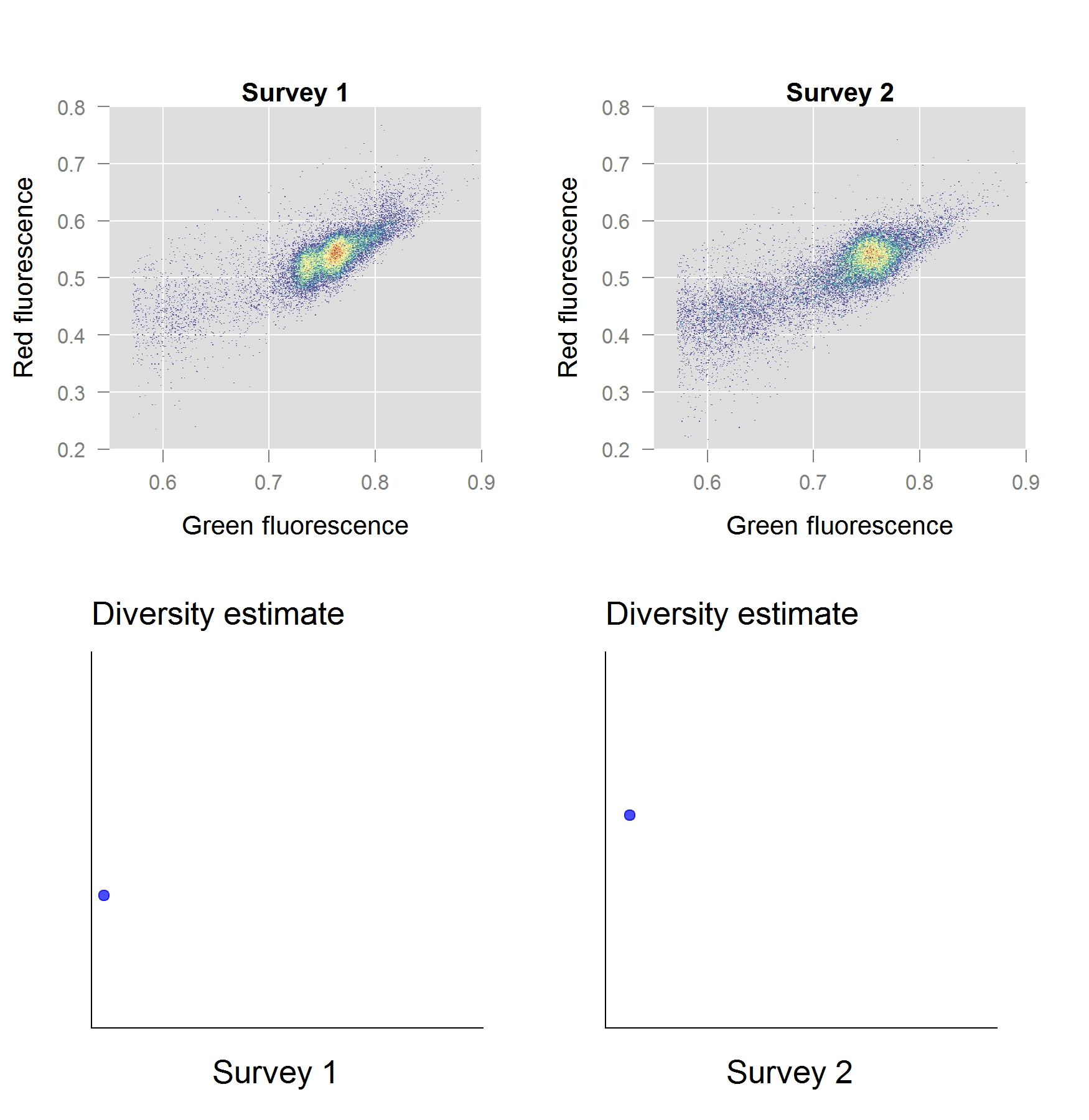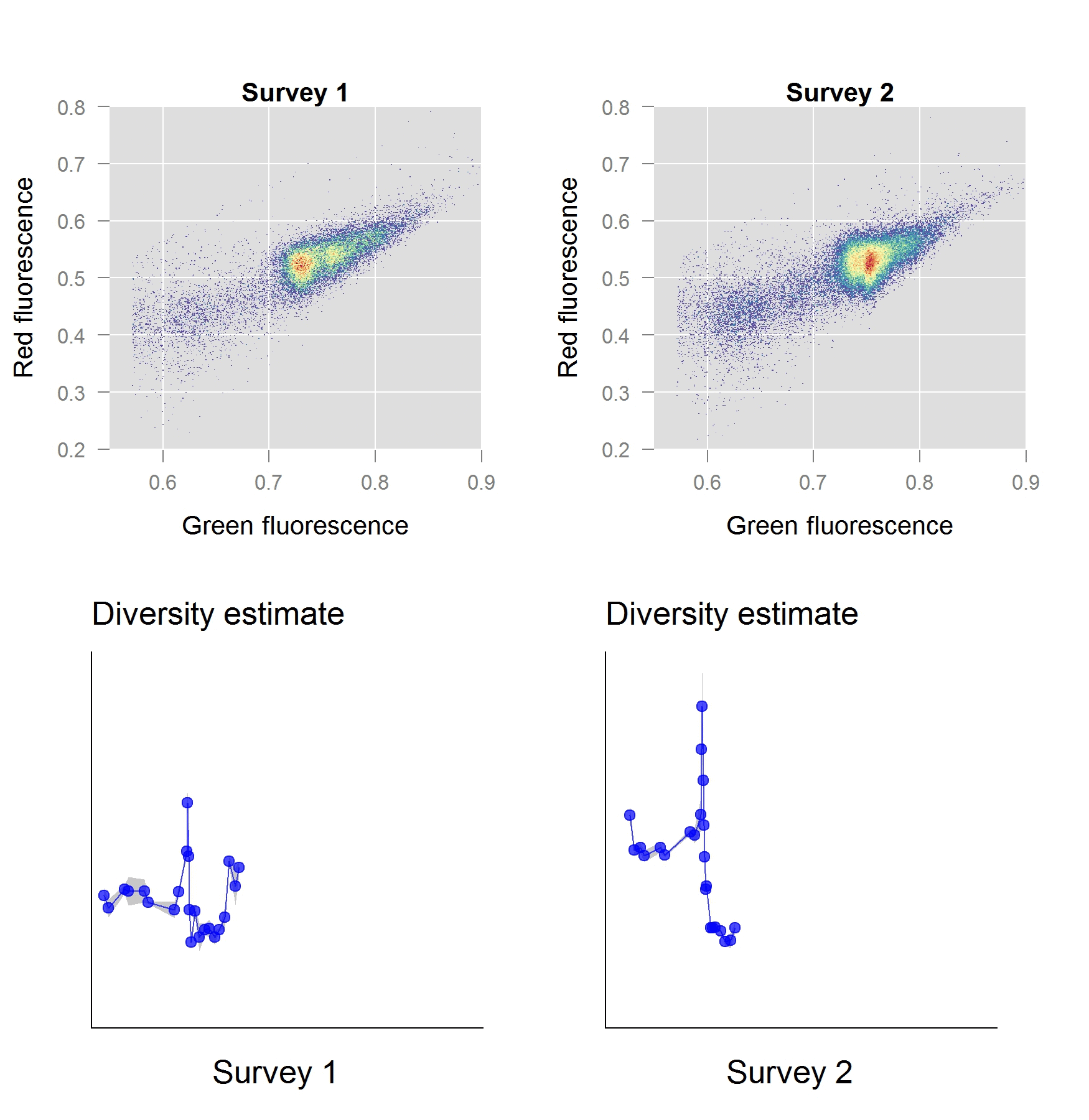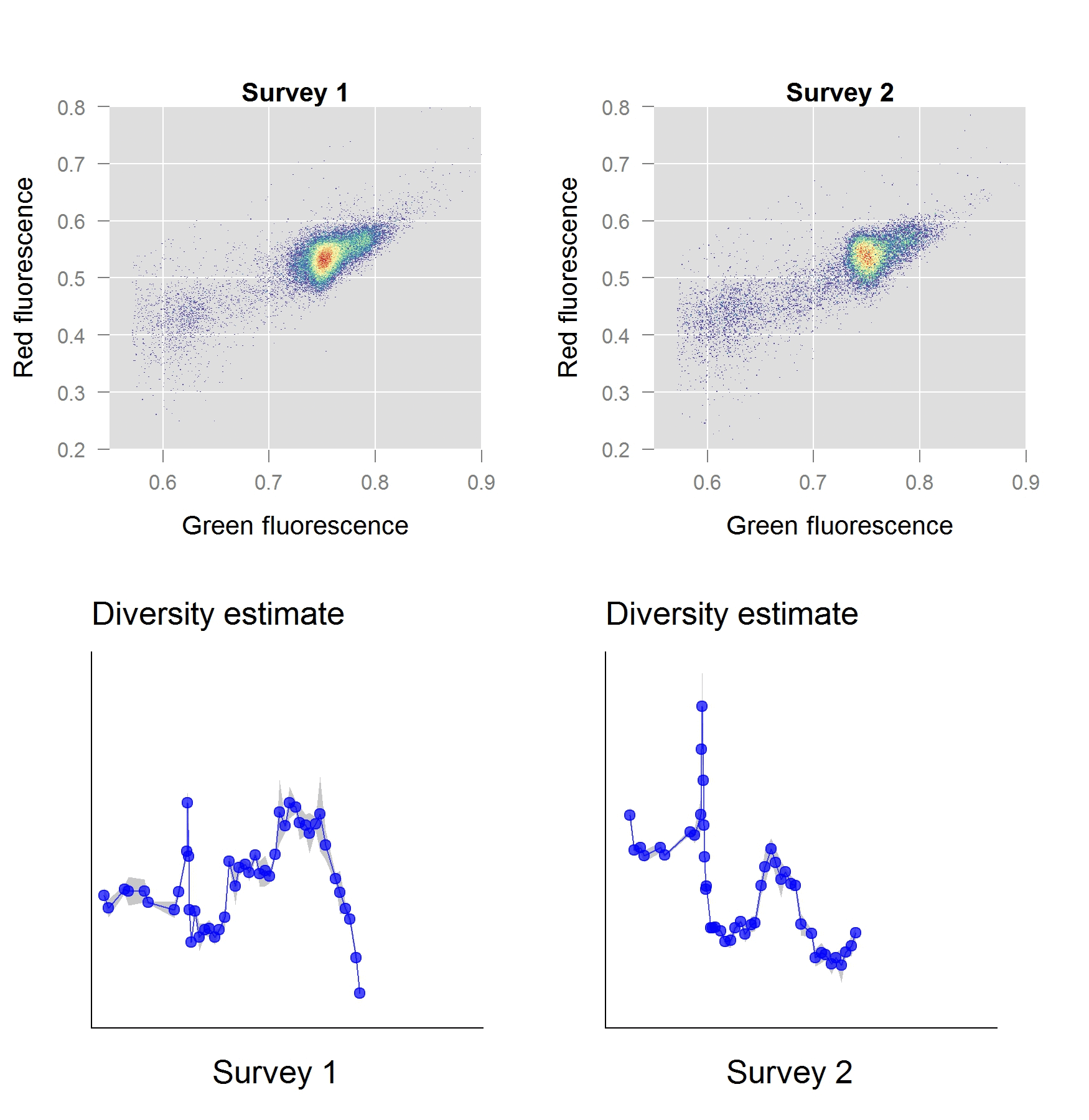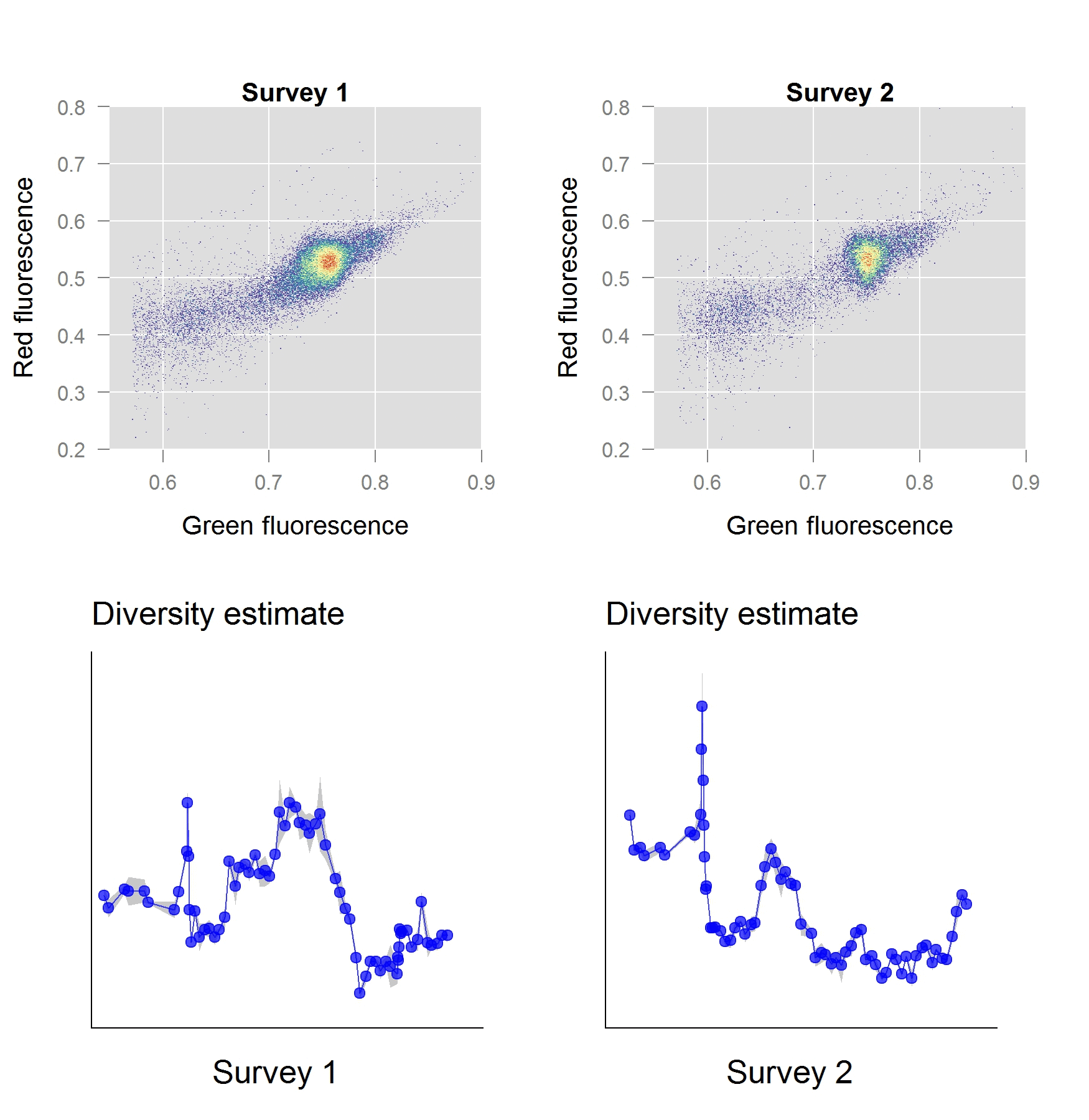
The Way Forward
Does all of this mean that phenotypic diversity metrics can completely replace the sequence-based methods? No, it doesn’t. Marker gene sequences harbour crucial taxonomic information, and in theory, should provide a higher resolution of the diversity. A tandem of both approaches will most likely prove to be the most powerful strategy moving forward.
A diversity estimate by flow cytometry is possible in minutes, with a sample volume of less than a millilitre, as opposed to the numerous litres which are often needed for a sequence-based analysis. The method is also straightforward, open-source and fast. Combine these attributes with the existence of portable, automated and on-site flow cytometers, and you’ve got a perfect environmental monitoring tool at your disposal.
As we’ve demonstrated in our paper, it’s easy to quickly identify samples of interest by the phenotypic diversity, and then further dissect the diversity through sequence-based analysis. This supervised method of data acquisition could open up new opportunities for research on long-term biodiversity dynamics in aquatic ecosystems.
But there is still work to be done as validation of the performance of these metrics is a necessary requirement for each ecosystem. While the phenotypic diversity metrics do not provide a one-stop shop, we must not forget that not all ecological variation, particularly at fine taxonomic scales, can be determined from marker genes, so alternative approaches for estimating microbial diversity are necessary to improve existing censuses. Once extensive validation for individual ecosystems has been attained, we hope that the phenotypic diversity metrics may become part of the standardized toolset for characterizing microbial diversity.
To find out more about phenotypic diversity measurements, read our Methods in Ecology and Evolution article ‘Measuring the biodiversity of microbial communities by flow cytometry’.

One thought on “Exploring Microbial Diversity: From the Sequence to the Cell”