Post provided by SILAS BOSSERT

Sequencing ultraconserved DNA for phylogenetic research is a hot topic in evolution right now. As the name implies, Ultraconserved Elements (UCEs) are regions of the genome that are nearly identical among distantly related organisms. They can provide useful information for difficult phylogenetic questions. The list of advantages is long – among others, UCEs are:
- easy to capture and sequence
- able to be retained from old and degraded museum material
- phylogenetically informative on different timescales.
A key reason for the method’s success is the developers’ commitment to full transparency, active tutoring, and willingness to help next-gen sequencing newbies like me to get started. Help is just a github-issue post away.
It took little to convince me that I wanted to use UCEs to reconstruct the phylogeny of one of my favourite groups of bees. I eventually met that objective but it won’t be part of this post. This blog post is about the journey to get there—the background story to the article ‘On the universality of target‐enrichment baits for phylogenomic research’.
Sequencing whose UCEs?
After spending two weeks in the Smithsonian Laboratory of Analytical Biology (and a little bit of waiting), we received and assembled our Illumina reads. These were exciting times, as our sequencing statistics turned out good and the contig lengths were promising. Often you get back a few particularly large contigs in the range of ~15-20 kilobases. Ideally these are large chunks, or even the entire mitochondrial genome, of your sample. This was the case for our reads. Intrigued by this by-catch, I started BLASTing these reads to determine what they match to.

I didn’t know what I was getting into. I anticipated sequencing bees related to honeybees and bumblebees, and ended up with a decent part of the second human chromosome as the longest contig—I assumed this was my own. What seemed to be a case of library contamination with nonspecific DNA became even more puzzling. I isolated the human contigs and searched them for UCEs of Hymenoptera (my group of interest). And … there were some ‘Hymenoptera UCEs’ in humans! In other words, there were sequences in these human contigs that were so similar to hymenopteran UCEs, that they were identified as such.
UCE Bait Sets
UCE baits – or the in-solution fishing rod that binds your target DNA and allows to pull it out of the genome-pond – are tailored for your group of interest. In my case this was Hymenoptera, a large group of insects including ants, bees, a variety of different wasps, and sawflies. The baits are generated by identifying shared conserved regions between sequenced genomes of your targeted group. If these regions are conserved among most all of the genomes, it is likely that other species in your group of interest may have these UCEs as well. This way you can identify UCEs in organisms that have never been sequenced before. There’s a great online tutorial that explains how to design baits yourself and a number of taxon groups have had baits generated using this workflow.
UCEs are conserved among bees, wasps, and beyond. We assume that they must undergo purifying selection to stay ‘as is’, which is exciting as many of these sequences are not characterised and their function isn’t yet understood. This opens new avenues of research, but also leads to the question: How widely shared are these UCEs? In our study, we address exactly this question.
.jpg)
We examined UCE baits which had been developed for different groups of arthropods, such as Hymenoptera, true bugs, or arachnids. We then screened all 242 publicly available arthropod genomes (as of early 2018) for sequences that match to these UCEs. Also, we included genomes of organisms that could in theory come in contact with our collection of insect specimens: humans, other mammals and a variety of different fungi and bacteria, totalling 400 genomes. Our screening procedures are reproducible with the code that we provide on GitHub. But be aware—our data volume was so conspicuously large that our university’s IT department felt the need to check in on us to figure out what we were doing!
Some UCEs are Particularly Widely Shared across the Tree of Life
We anticipated little and found a lot. Hundreds of UCEs are shared across the whole of the insect Tree of Life and beyond. Even humans have nearly 100 regions that seem like they are the same (or at least very similar) sequences to Hymenoptera UCEs. It’s amazing that these lineages that split in very deep time, presumably over 500 million years ago, have genomic elements that remained nearly unchanged. Our findings show that we can take advantage of these patterns and harness the universality of these baits. A single bait set can capture putative orthologous sequences from the entirety of the most diverse extant phylum Arthropoda, encompassing over 1,200,000 described taxa and about 80% of all known species of animals.
We can also expand our knowledge to understand UCEs and the biology of most essential cellular processes. The 100 most widely shared UCEs (based on the Hemiptera bait set) code for tasks that seem indispensable for life as we know it. They include transcription factors, motor proteins, membrane receptors and more. But this intriguing biology of very conserved DNA comes with the reminder that we have to work carefully in the lab and thoroughly assess our sequence data. This is particularly relevant for organisms with predatory feeding behaviours and potential contamination on appendages that are used to capture prey. Some insects may even have internal insect parasitoids or ectoparasites that are easily overlooked but might very well contribute DNA to your extraction buffer.
Overcoming Risks of Contamination
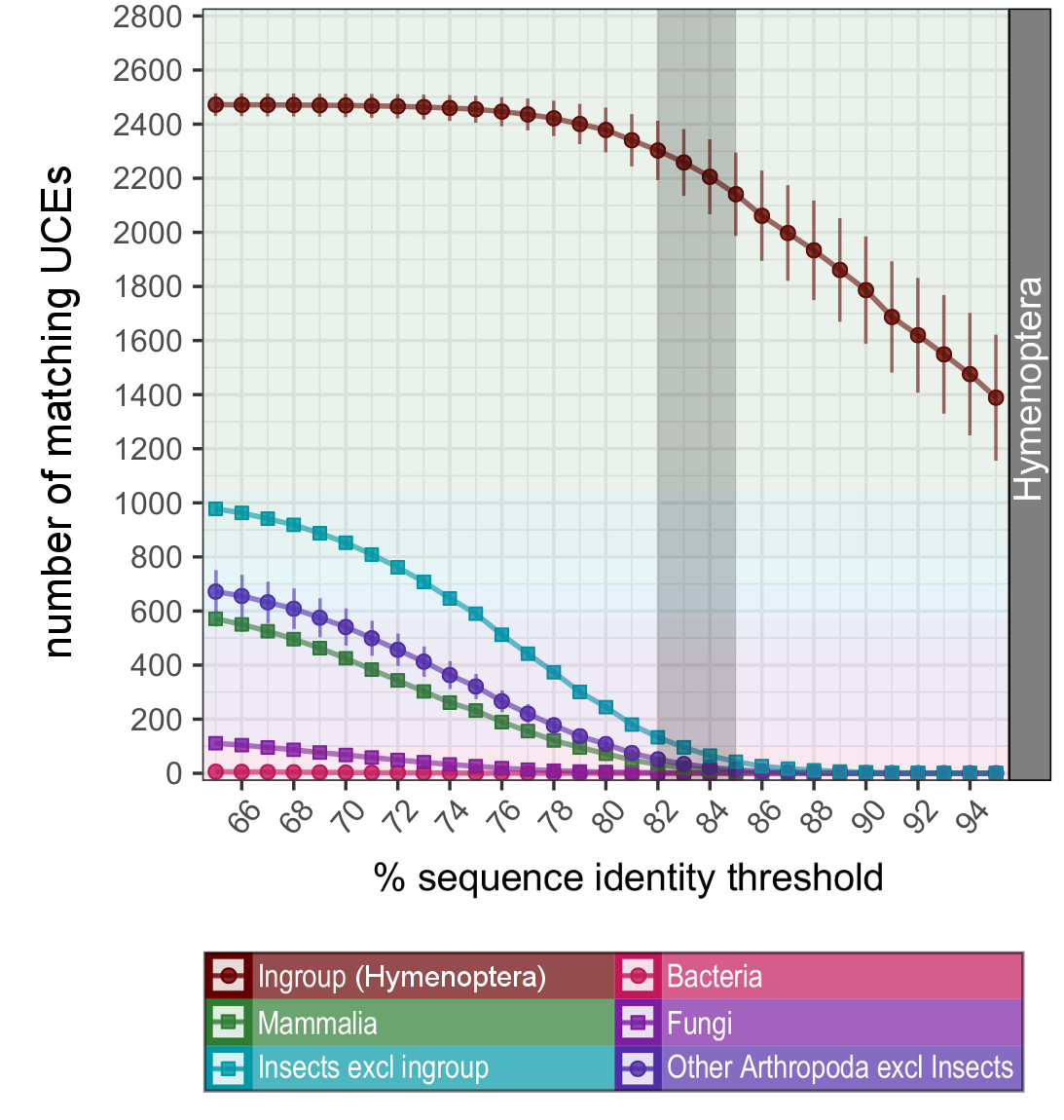
Fortunately, it’s straight-forward to bioinformatically remove contaminants from organisms other than your group of interest, even if accidentally sequenced. We can tighten a screw in our parameters to identify UCEs during the bioinformatic processing. In the identification step, the UCE baits are matched in-silico against all sequencing reads, in order to fish out UCEs from the pool of everything that was sequenced. If we force this search to be really strict, you can separate the wheat from the chaff—retain your ingroup’s DNA and discard contaminants. We also present some other tricks that might help to identify contamination in the first place. This quality control is certainly worth the time and finally helped me to make progress on my initial project—while I rest assured that I am not part of the taxon sampling.
To find out more, read our Methods in Ecology and Evolution article ‘On the universality of target‐enrichment baits for phylogenomic research’.
This article is part of the Virtual Issue ‘Phylogenetics and Comparative Methods: The Dark and Bright Sides’.
