Post provided by Jordan Cuff and Lorna Drake

The cover of our March issue shows a female Eurasian otter (Lutra lutra), with its young kit bringing a crab to shore before eating it. The photographer, Alan Seymour, had been watching the otter from a distance, while laying low behind a large boulder. Direct observations of trophic interactions take a lot of time and skill, especially concerning cryptic species (e.g., semi-aquatic mammals, small invertebrates). Accurately and precisely identifying prey items is mostly impossible by observation. Molecular methods, such as DNA metabarcoding, offer a sensitive approach for investigating trophic interactions of cryptic species; however, this high sensitivity can introduce errors. In this post, the authors discuss potential sources of errors in dietary metabarcoding datasets, and how to use minimum sequence copy thresholds to carefully remove them, detailed in their Methods in Ecology and Evolution article “An assessment of minimum sequence copy thresholds for identifying and reducing the prevalence of artefacts in dietary metabarcoding data”.
When to be negative about positives: false positives in metabarcoding
DNA metabarcoding has drastically increased our ability to analyse the diets of animals in the wild. The main benefit of DNA metabarcoding is its extreme sensitivity, but this is subject to some critical trade-offs. Most notably, DNA metabarcoding is prone to detecting false positives. False positives can occur via environmental or lab contamination, and even sequencing errors, resulting in DNA artefacts. These phantom presences can be obvious (e.g., human DNA within tiny farmland money spiders’ stomachs) but are often quite problematic (e.g., perfectly reasonable prey DNA transferred between samples), making their removal all the more difficult.
Taking out the trash
To deal with these problems, molecular ecologists alike have often turned to using minimum sequence copy thresholds that remove detections represented by low read counts. . The nature of these thresholds is variable however, with many studies simply using one or ten reads as the cut-off across the whole dataset, which doesn’t account for differences in total read depth between samples, nor differences between the total read depth of different studies. This leads to the overzealous removal of legitimate data and, simultaneously, false positives remain in the dataset. An alternative method involves removing reads below a certain percentage of the total read count of each sample, but the performance of this approach hasn’t been extensively compared with real-world data.

A tale of eight methods: a comparison of minimum sequence copy thresholds
In our study, we compared a variety of minimum sequence copy thresholds and the effectiveness of these thresholds when used in combination against four sequencing runs from two datasets: one concerning dietary variation in otters across Britain, and the other focused on the gut contents of spiders in barley crops. The minimum sequence copy thresholds assessed included:

- Arbitrary (removing read counts less than one or ten)
- Negative control-based (removing reads equal to or less than those in negative controls per taxon)
- Sample-based (reads below a percentage of read depth per sample)
- Sequencing run-based (reads below a percentage of the total read depth)
- Taxon-based (reads below a percentage of read depth per taxon)
The methods performed very differently, shifting the balance of false positives and negative accordingly. Some methods were very clearly overly conservative, while others left behind known false positives.
Our optimum, but is it yours?
We ultimately that the most effective method of balancing presence of false positives and negatives in the data was the combination of a negative control-based threshold alongside a fine-tuned percentage of sample read count threshold. Whilst a negative control-based threshold is easily defined as the maximum read count identified in negative controls per taxon (i.e., amount of sequenced DNA in negative controls), choosing a percentage sample threshold is more difficult as it is dependent on identifying occurrences in your dataset that are known to be false positives.
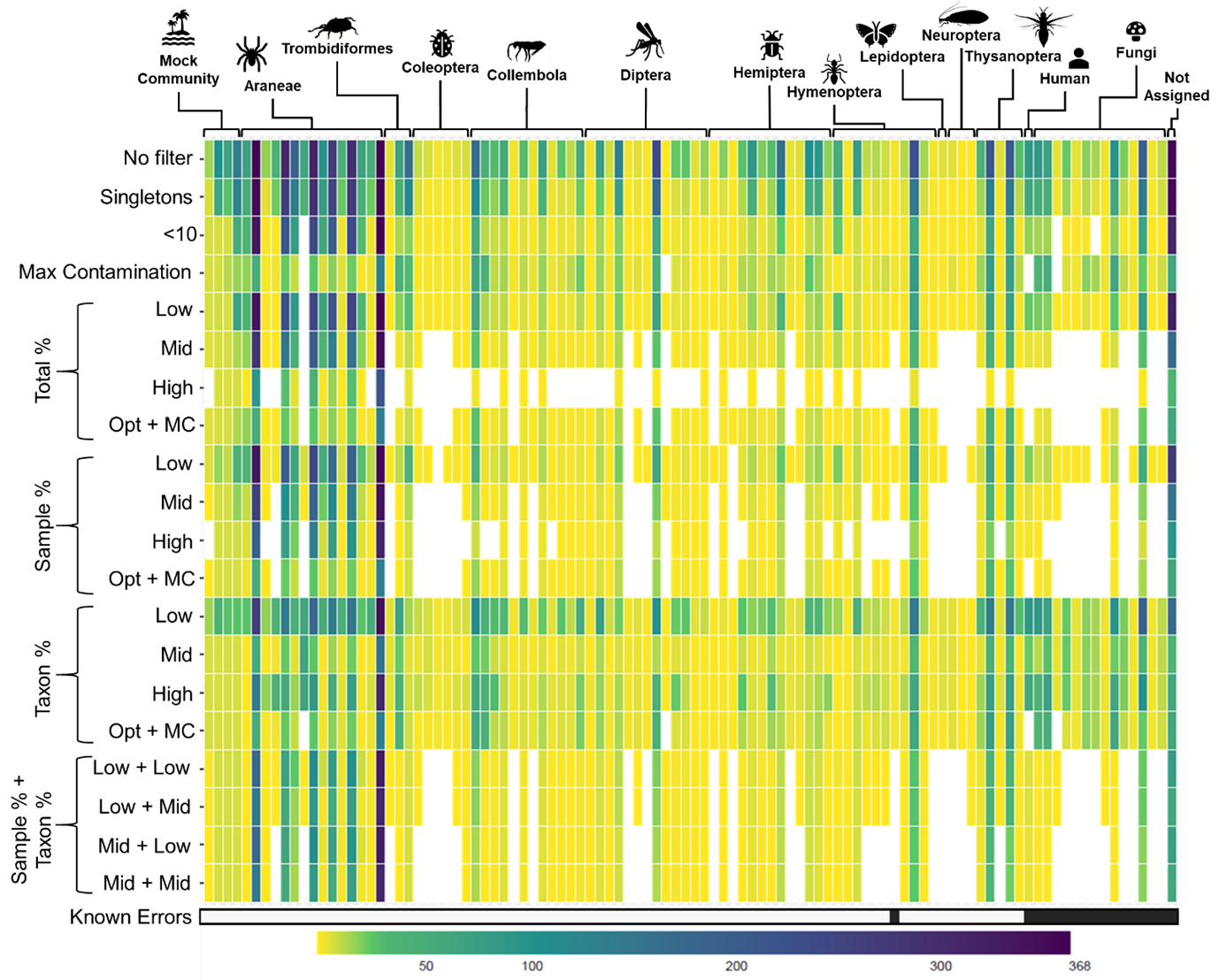
While we found this approach effective, but that’s not to say it will be for every DNA metabarcoding study. Refining and adjusting thresholds on a study-by -study basis is advised, as factors such as sequencing depth, diet of the focal species and the extent of ‘host’ predator amplification will vary significantly. Finally, the thresholds applied clearly have a massive impact on the resulting DNA metabarcoding dataset, therefore it is important for researchers to report the thresholds used for transparency and reproducibility.
To read the full study by Drake et al., “An assessment of minimum sequence copy thresholds for identifying and reducing the prevalence of artefacts in dietary metabarcoding data”, visit the Methods in Ecology and Evolution journal website here.
