Post provided by KAYLEIGH O’KEEFFE

Pathogens and the infectious diseases that they cause can have devastating impacts on host individuals and populations. To better understand how pathogens are able to cause disease, we can investigate the genetic mechanisms underlying the infection process. Hosts may respond to infection by upregulating defence pathways. Pathogens, in turn, evade these host immune responses as they infect and cause disease. As this process unfolds and each organism responds to the other, gene expression changes in both the host and the pathogen. These gene expression changes can be captured by dual RNA‐seq, which simultaneously captures the gene expression profiles of a host and of a pathogen during infection.
The Problem with Alignments of Sequencing from Non-Model Host/Pathogen Systems
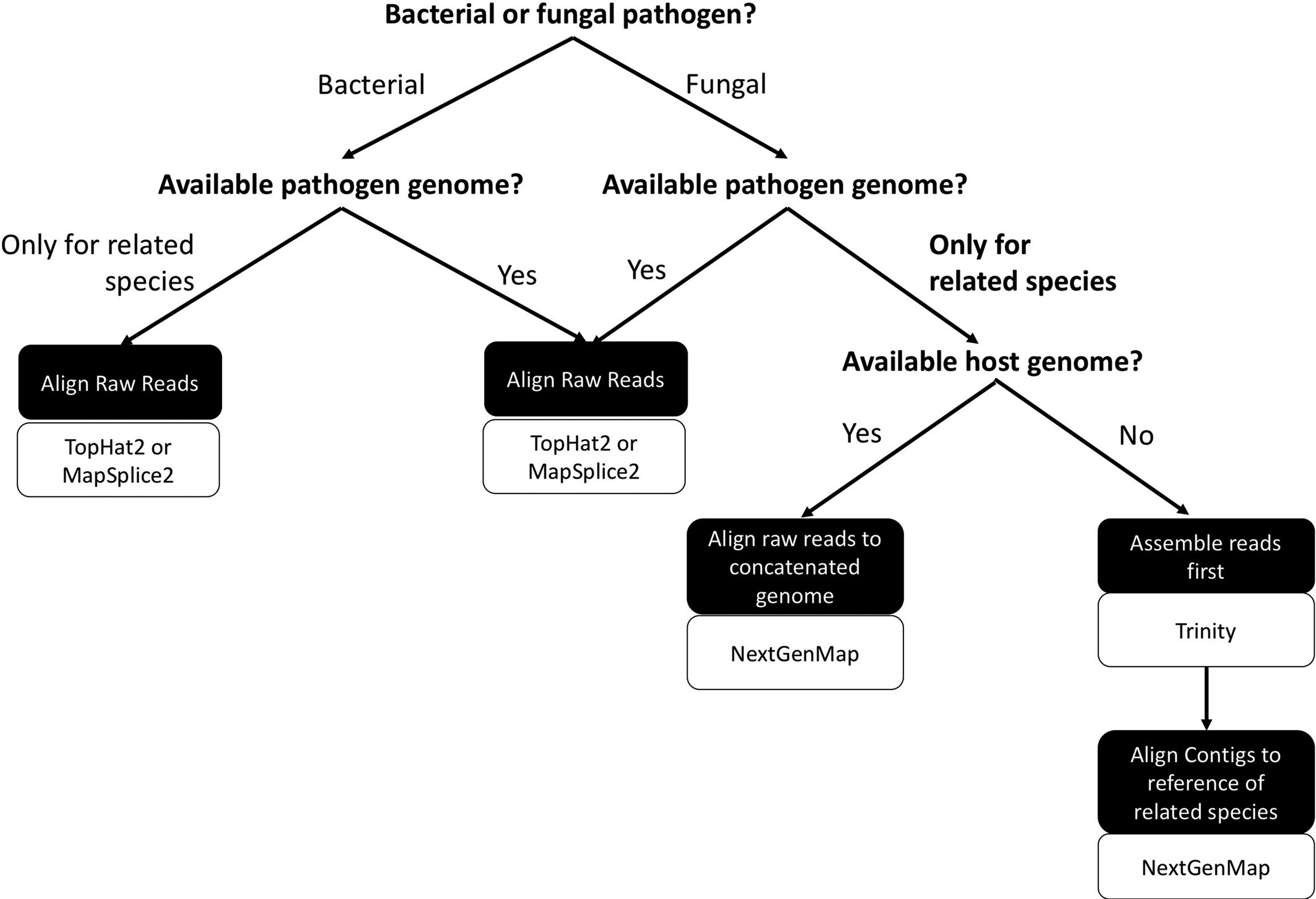
Dual RNA-seq involves sequencing host tissue that is infected with a pathogen, so the sequencing is always a mixture of reads from the host and reads from the pathogen. The traditional approach to analyzing this type of data involves sorting the reads apart by mapping them to the genomes of the pathogen and the host. But what if you’re working with a system where genomic resources are limited?
Only a small fraction of species – which we call model species – have sequenced and annotated reference genomes. This means that species within many host/pathogen systems have limited genomic resources. I’m a disease ecologist, and the plant host and fungal pathogen that my empirical work focuses on (pictured above) both have limited genomic resources. I was therefore very motivated to investigate if and how dual RNA-seq could be used in non-model systems like my own. After discussions with Corbin Jones, an evolutionary geneticist at UNC-Chapel Hill, we started investigating the accuracy of different approaches to analyzing dual RNA-seq when genomic resources with sequencing datasets that we simulated.
Does it Help to Have a Genome for a Closely-Related Species?
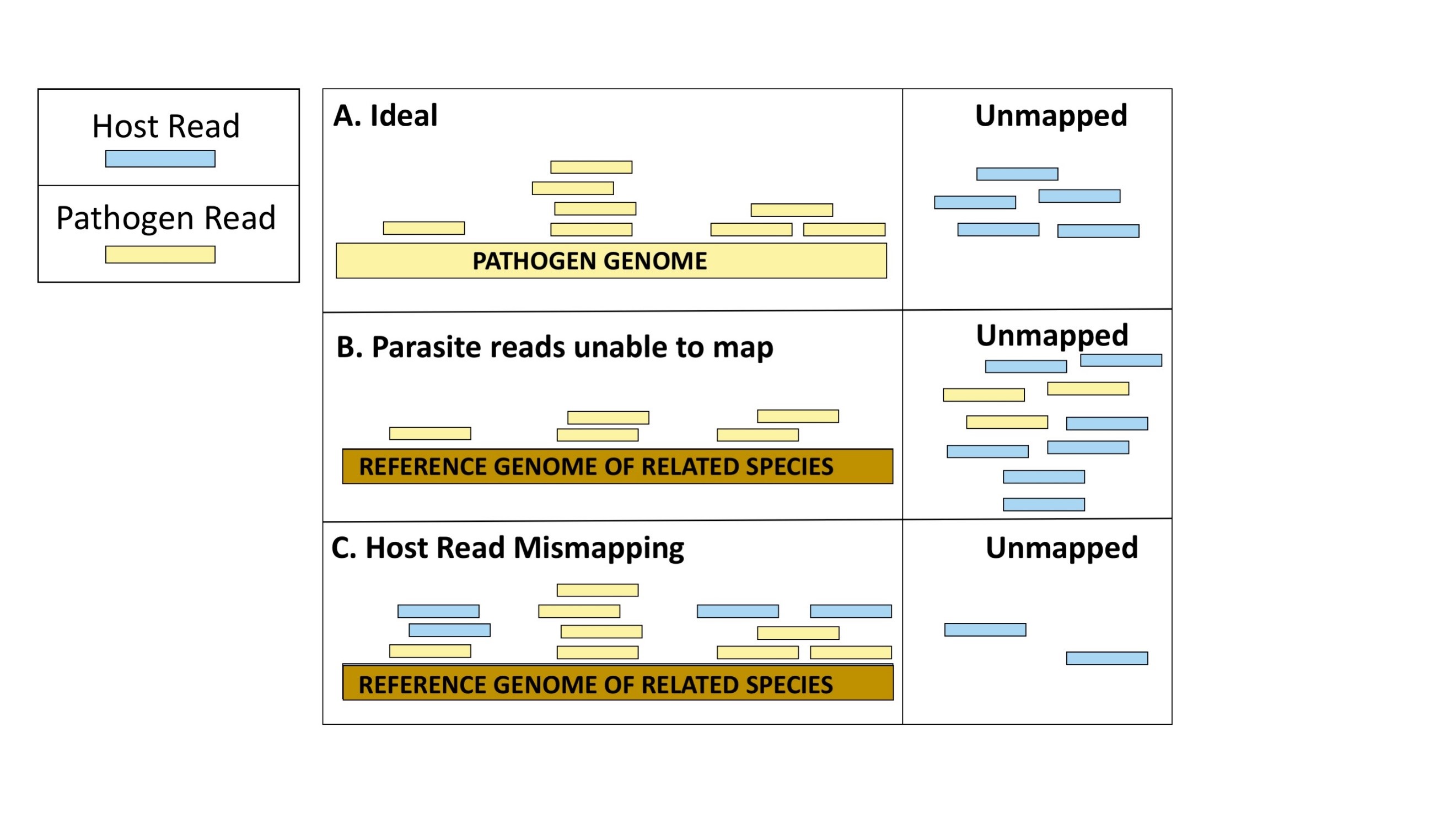
For non‐model organisms, traditional approaches to analyzing RNA‐seq data include mapping reads to reference genomes of related species. Using dual RNA-seq datasets that we simulated of a plant and fungal pathogen system, we first investigated whether you could use the genome of a species closely related to the pathogen to align pathogen reads from these sequencing mixtures. We found that aligners that were able to map pathogen reads to these related genomes also mismapped reads that actually originated from the host. This would have huge implications for interpretations of this data, as regions where mismapping occurs would be quantified as overexpressed.
What if You Had a Host Genome?
We then investigated approaches to mitigate this read mismapping. What if, in addition to the genome of a species closely-related to the pathogen, the host genome was available? We concatenated (the technical term for ‘linked together’) the genomes of the host and species closely-related to the pathogen and performed alignments with this composite genome as the reference in a method we called the ‘place-to-go’ design. Mapping dual RNA-seq to such a concatenated genome resulted in substantially fewer host reads mismapping to the genome of the species closely-related to the pathogen. It also retained the ability map pathogen reads.
What if You Don’t Have a Genome for the Pathogen OR the Host?
We also looked into whether first assembling reads de novo into longer fragments could reduce host reads from mismapping to the genome of a species closely-related to the pathogen. We found that this also substantially reduced host read mismapping while retaining the ability to map pathogen reads.
Suggestions for Future Applications
Host reads mismapping to the genome of the pathogen (or the genome of a species closely related to the pathogen) can have severe implications for the characterization of the gene expression profile of the pathogen during the infection process. Furthermore, while we were able to identify the species of origin (host or pathogen) of reads within our simulated sequencing, the species of origin of the reads from real dual RNA-seq would not be able to be definitively identified. This could result in such inaccuracies going unnoticed, and not taking measures to avoid reads from mismapping can lead to inaccurate genetic mechanisms implicated in the infection process.
Our systematic comparison of analytical approaches to using dual RNA-seq in non-model host-pathogen systems took advantage of simulated sequencing. We suggest a workflow to follow to determine the best approaches to extending the use of dual RNA‐seq to a wider array of systems.
As genomic technologies and their analytical tools continue to become more affordable and accessible, it’s important to critically assess how accurate genomic analyses with these tools are. Furthermore, as infectious diseases are expected to have increasingly detrimental impacts in the coming years, it’s critical that we investigate proper methods of analyses to ensure accurate insights are gained as systems are explored.
To find out more, read our Methods in Ecology and Evolution article ‘Challenges and solutions for analysing dual RNA‐seq data for non‐model host–pathogen systems’

One thought on “Using Dual RNA-seq to Investigate Host-Pathogen Systems When Genomic Resources are Limited”