Fossil of a Horseshoe crab (Museum of Natural History Berlin)
A couple of days ago I came across a nice video (in Portuguese only, sorry) about so-called “living fossils”. The video focused on the problems of using them as arguments against evolution. But I’d like to take the opportunity to talk more about these long-lived lineages.
‘Living fossil’ is a term used to describe lineages that are thought to have been around for a very long time and retain characteristics that resemble of their fossil relatives. A couple of well-known examples of these lineages are the Tuatara of New Zealand (Sphenodon punctatus) and the Gingko tree (Gingko biloba).
The latest issue of Methods in Ecology and Evolution is now online! This month’s issue is a little shorter than our last few. But, as they say, good things come in small packages!
Executive Editor Aaron Ellison has selected six Featured Articles this month. You can find out about all of them below. We’ve also got five Applications articles in the March issue that we’re going to cover.
Sequencing ultraconserved DNA for phylogenetic research is a hot topic in evolution right now. As the name implies, Ultraconserved Elements (UCEs) are regions of the genome that are nearly identical among distantly related organisms. They can provide useful information for difficult phylogenetic questions. The list of advantages is long – among others, UCEs are:
phylogenetically informative on different timescales.
A key reason for the method’s success is the developers’ commitment to full transparency, active tutoring, and willingness to help next-gen sequencing newbies like me to get started. Help is just a github-issue post away.
Five years ago at Evolution 2014, ‘The Dark Side of Phylogenetics’ symposium (organised by Natalie Cooper) explored some of the issues with phylogenetic comparative methods (PCMs). This year at Evolution 2019, Michael Landis and Rosana Zenil-Ferguson are organising a contrasting ‘Bright Side of Phylogenetics‘ spotlight session (featuring Michael Matschiner). They aim to promote research that has overcome these pitfalls and that explores innovations in phylogenetics. Clearly they found our lack of faith disturbing.
Researchers from Canada and the USA found that tree and shrub genetics can be used to produce more accurate predictions of when leaves will burst bud in the spring. Their study was published in Methods in Ecology and Evolution.
Although climate sceptics might find it hard to believe with this year’s endless snow and freezing temperatures, climate change is making warm, sunny early springs increasingly common. And that affects when trees start to leaf out. But how much?
Plant-pollinator interactions are often considered to be the textbook example of co-evolution. But specialised interactions between plants and pollinators are the exception, not the rule. Plants tend to be visited by many different putative pollinator species, and pollinating insects tend to visit many plant hosts. This means that diffuse co-evolution is a much more likely driver of speciation in these communities. So, the standard phylogenetic methods for evaluating co-evolution aren’t applicable in most plant-pollinator interactions. Also, many plant-pollinator communities involve insect species for which we do not yet have fully resolved phylogenies. Continue reading “2018 Robert May Prize Winner: Laura Russo”
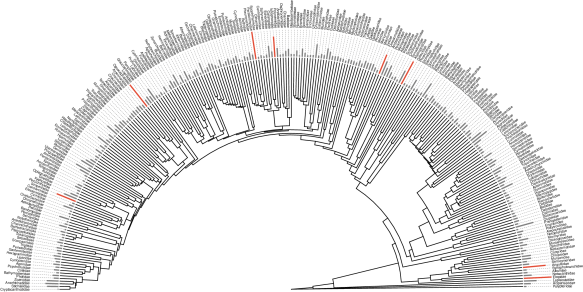
In our recent publication (Rabosky et al. 2018) we assembled a huge phylogeny of ray-finned fishes: the most comprehensive to date! While all of our data are accessible via Dryad, we felt like we could go the extra mile to make it easy to repurpose and reuse our work. I’m pleased to report that this effort has resulted in two resources for the community: the Fish Tree of Life website, and the fishtree R package. The package is available on CRAN now, and you can install it with:
install.packages("fishtree")
The source is on GitHub in the repository jonchang/fishtree. The manuscript describing these resources has been published in Methods in Ecology and Evolution (Chang et al. 2019).
Analyzing diversification rate heterogeneity across phylogenies allows us to explore all manner of questions, including why Australia has such an incredible diversity of lizards and snakes.
Within the tree of life there are differences in speciation and extinction rates over time and across lineages. Biologists have long been interested in how speciation rates change as a function of ecological opportunity or whether key innovations lead to increases in the rate of speciation. Exploring this rate variation and examining how clades differ in terms of their diversification dynamics can help us to understand why species diversity varies so dramatically in time and space. Learning more about the relationship between traits and diversification rates is especially important because it has the potential to reveal the causes of pervasive variation in species richness among clades and across geographic regions.
Today is the first day of the Crossing the Palaeontological-Ecological Gap (CPEG) conference. The aim of the conference is to open a dialogue between palaeontologists and ecologists who work on similar questions but across vastly different timescales. This splitting of temporal scales tends to make communication, data integration and synthesis in ecology harder. A lot of this comes from the fact that palaeontologists and ecologists tend to publish in different journals and attend different meetings.